
 |
|
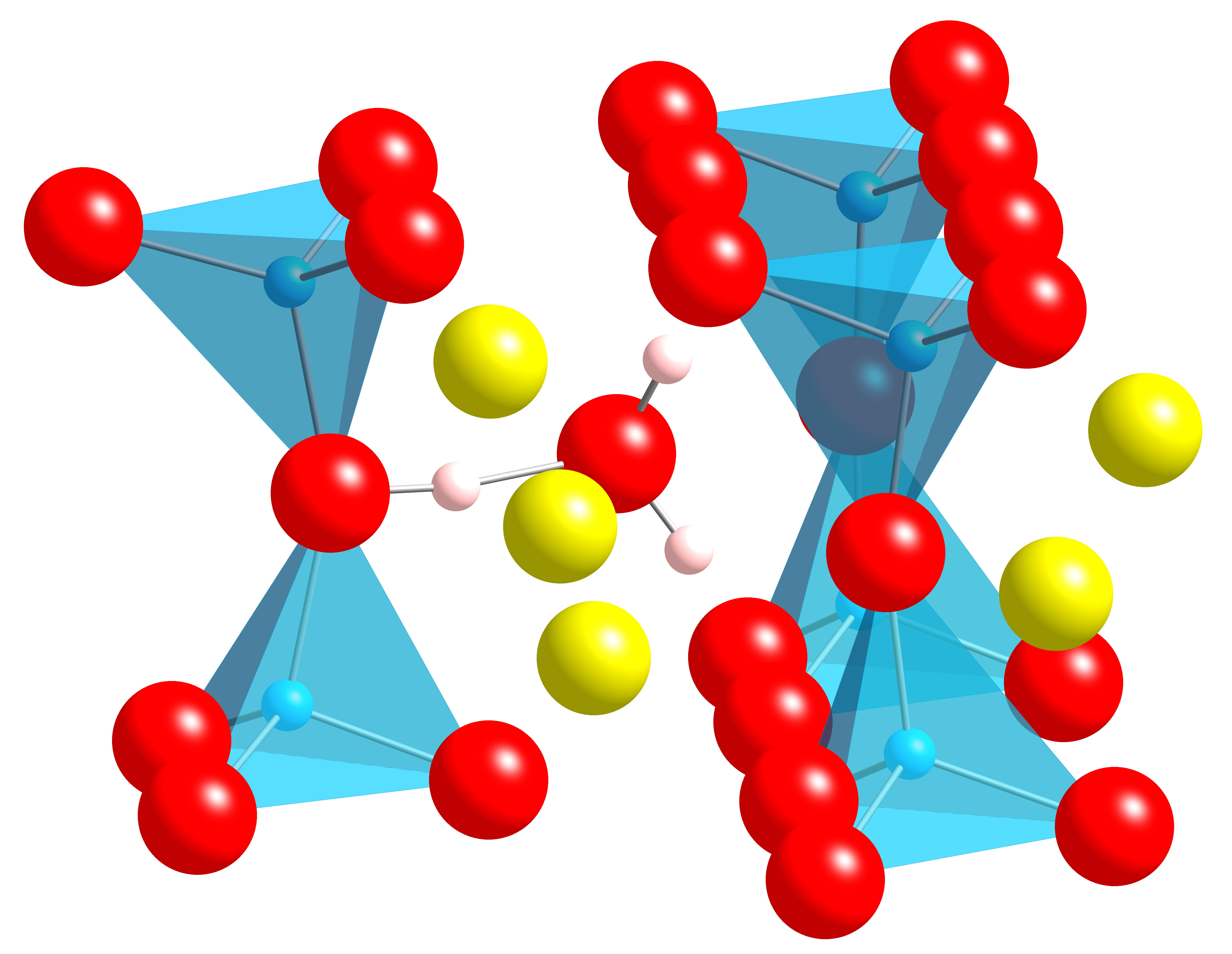
Figure 1 Structure of hydronium molecule |
Professor Alastair Cormack’s group at the New York State College of Ceramics at Alfred University used the Handy cluster at IACS for two sets of calculations. Both used the VASP code to perform quantum mechanical simulations.
The first project was concerned with the simulation of water in sodium beta”-alumina. Aging effects (that is, a reduction ionic conductivity with time when the material is stored under ambient conditions) are thought to be related to the uptake of water by the beta”-alumina. In this work, the structure of protons and hydronium ions in the conduction plane was examined, when these species are exchanged with sodium in the conduction plane. The most stable configuration for a single hydronium is shown in Figure 1. Using MD, it was further shown that the presence of the water molecule had a deleterious effect on the sodium mobility. The presence of the protonic species required the use of density functional theory (DFT); this material has a very large unit cell, which makes the calculations challenging, making use of computational resources like Handy essential.
 |
|
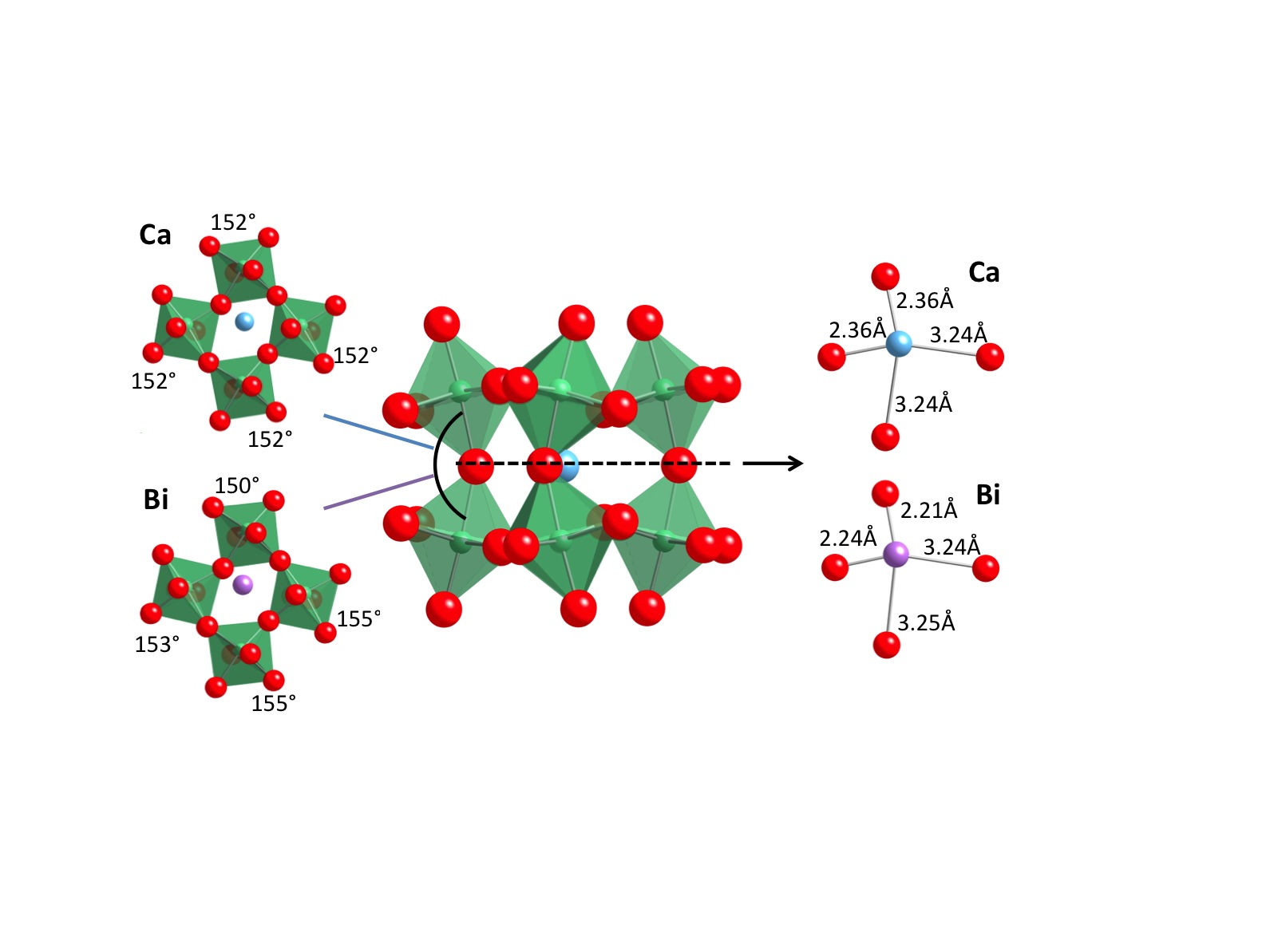
Figure 2 Octahedral distortions |
The second project concerned a hybrid functional study of cation disordering in Bi2CaNb2O9 (BCNN), which belongs to the Aurivillius family of layered perovskites. It is one of the more important classes of photocatalysts and has shown remarkable efficiency in splitting water into hydrogen and oxygen. To understand and enhance its photocatalytic activity, the effect on its electronic structure, due to subtle lattice distortions caused by cation site-mixing (see Figure 2), was studied. The availability of the Handy cluster allowed us to investigate the problem with sophisticated hybrid-functional ab-initio simulations. To our knowledge, this is by far the largest oxide system investigated using such methods. The results showed improved agreement with both diffraction experiments and band gap measurements, compared with the DFT method. It also revealed that cation site-mixing plays a critical role in the band gap related properties, which may be used to further engineer the material.
![]()
PH: 631-632-4629 | FX: 631-632-4125 | iacs@stonybrook.edu