
A team of international scientists led by Stony Brook University researchers have created an ultra-fast, computerized way to model protein interactions, potentially helping to speed needed drugs to market.
 |
Dima Kozakov, PhD, lead author and Assistant Professor in the Department of Applied Mathematics and Statistics |
Protein-protein interactions (PPIs) are the basis of cellular functions, and when these processes are compromised diseases such as cancer emerge. For years scientists have tried with mixed success to map out PPIs to understand cellular processes. Now researchers led by Stony Brook’s Dima Kozakov have outlined a method that could pave the way to designing new drugs that prevent problematic protein interactions that lead to disease. The findings are published in the early online edition of PNAS.
Proteins are the major building blocks of the cell. Many proteins perform their function by interacting with other proteins. In a typical cell, hundreds of thousands of different protein interactions take place. Characterizing the structure of these interactions helps elucidate how organisms function normally and during disease development.
“The problem considered is given three dimensional structures of two individual proteins to predict how these protein interact with each other,” said Kozakov, lead author and Assistant Professor in theDepartment of Applied Mathematics and Statistics in the College of Engineering and Applied Sciences, and a faculty member of the Laufer Center for Physical and Quantitative Biology and Institute for Advanced Computational Science (IACS) at Stony Brook University. He likened the method to characterizing all the possible structures that pairs of “lego blocks” form out of a huge set of different individual starting blocks.
In the paper titled “Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds,” the authors explain a new algorithm used to create ultra-fast approach to modeling protein interactions. They discovered that the method runs 10 to 100 times faster than previous state-of-the-art methods, without compromising accuracy.
The researchers used something called fast Manifold Fourier transform (FMFT) that help speed the calculations, enabling them to sample a large number of putative protein-protein complex conformations. 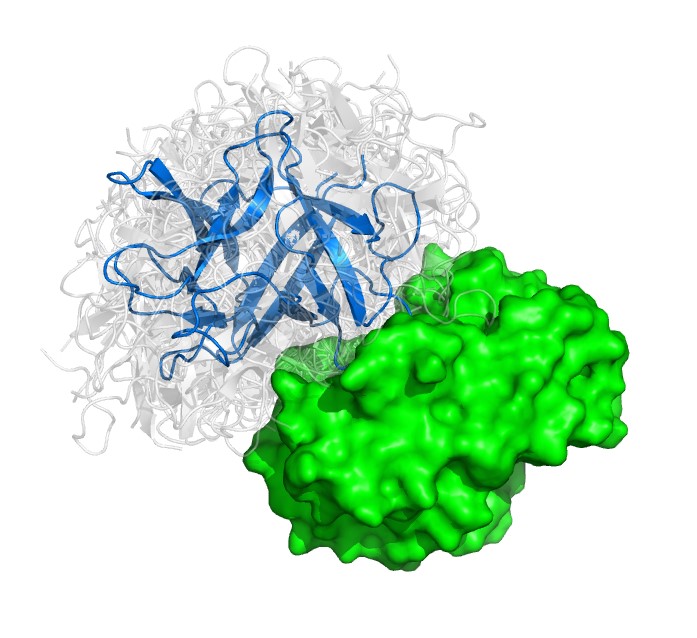
Correct structure of a protein-protein complex together with an ensemble of alternative structures sampled by the new algorithm.
“The idea behind the approach is to present proteins as a combination of quantum shapes that enable fast simultaneous analysis of many pairs using a single calculation rather than assessing each pair independently,” summarized Dr. Kozakov. “The approach can be run in under 15 minutes on a personal laptop and can be used instead of costly experimental techniques to determine the structure of the protein-protein complex.”
The new algorithm will soon be available to the scientific community through the publicly available protein-protein docking server called ClusPro. This resource, with more than 15000 academic users worldwide, supported by the National Science Foundation and the Binational Science Foundation, is being developed by the same research team at the Laufer Center and IACS, in collaboration with scientists at Boston University.
ClusPro was judged to be the best automated docking server in the latest rounds of the international blind protein docking competition called CAPRI (Critical Assessment of Prediction Interaction).
Dr. Kozakov and colleagues believe the new method, implemented in the ClusPro server, will enable scientists to model protein interactions for the whole cell, a key step to designing drugs that prevent defective protein interactions that lead to disease.